
什么是标准电极电势?
说明:标准电极电势1.23 V是析氧反应(OER)的热力学平衡值,源于水分解的吉布斯自由能变(ΔG°=237.2 kJ/mol),该值在标准条件(298 K, pH=0)下统一适用于酸/碱体系,但实际反应因多步质子–电子转移能垒需克服显著过电位(η)。
在应用中,1.23 V是评估催化剂活性(如η、Tafel斜率)的核心基准,指导电解槽设计与能量效率优化,同时需根据pH、温度及氧气分压进行能斯特修正。
标准电极电势的定义
标准电极电势(E∘)是指在标准条件(298 K、1 bar气体压力、1 mol/L溶液浓度)下,以标准氢电极(SHE,电势定义为0 V)为参考基准测得的半反应还原电势。对于析氧反应(OER),其标准电势1.23 V的推导源于热力学数据:
反应吉布斯自由能变(ΔG°):
水分解反应的总自由能变为ΔG°=237.2 kJ/mol(2H₂O→2H₂+O₂)。
OER半反应(酸性条件):
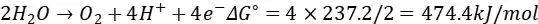
根据公式E∘=-ΔG∘/(nF)(n为转移电子数,F为法拉第常数),计算得:
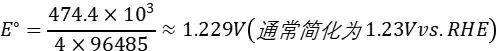
与pH无关的特性:
在碱性条件下,OER反应式为4OH–→O2+2H2O+4e–,但通过可逆氢电极(RHE)标度换算后,平衡电位仍为1.23 V。这是因为RHE已包含pH修正,使得不同pH下的热力学驱动力统一。
1.23V的实验测定与验证
电化学测量方法:
通过Debye-Hückel理论和不同离子强度下的电势插值法,测得OER标准电势为1.229 V vs. SHE;方式二:使用红氧电池(redox cell)结合金电极或汞电极,在标准条件下直接验证该值。
pH与温度的影响:
pH效应:根据能斯特方程
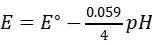
温度效应:温度系数为-0.846 mV/K(298 K基准)。例如:303 K时电势降至1.226 V。
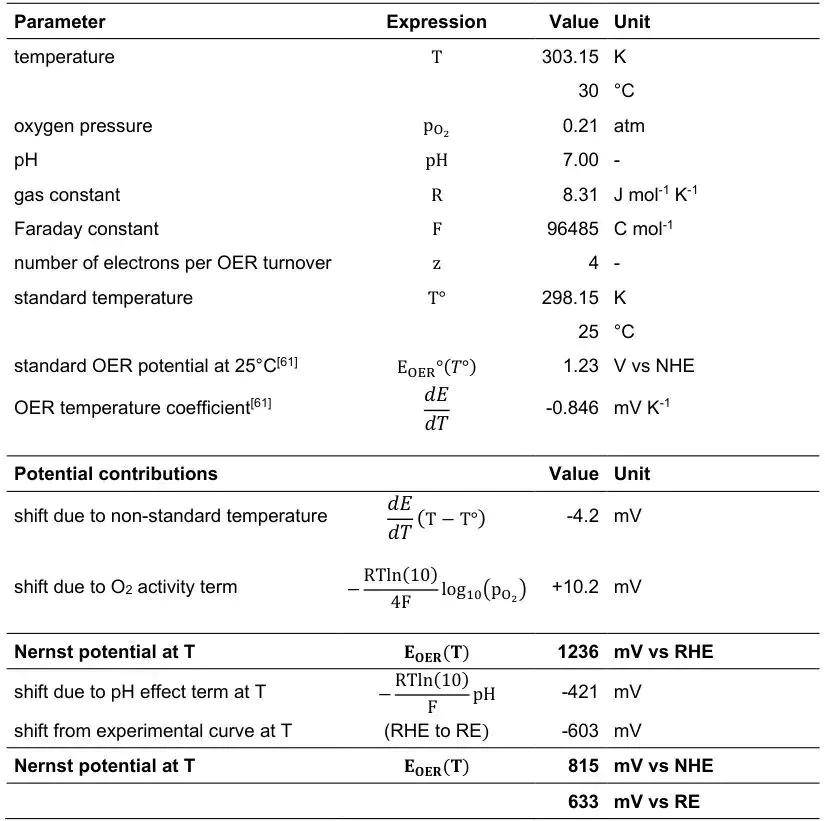
高温电解槽(>60°C)需重新计算实际平衡电位。
OER反应机理与动力学挑战
尽管热力学平衡电位为1.23 V,实际反应需克服多步能垒,导致显著过电位(η):
反应路径(以酸性条件为例):
步骤1:H2O+*→OH*+H++e–
步骤2:OH*→O*+H++e–
步骤3:O*+H2O→OOH*+H++e–
步骤4:OOH*→O2+H++e–+*
其中O*→OOH*步骤的吉布斯自由能变最高(~3.2 eV),成为速率决定步骤。
理论最小过电位:
理想催化剂应使各步骤能垒相等(即ΔG₁=ΔG₂=ΔG₃=ΔG₄=1.23 eV),但实际催化剂因吸附能差异需额外过电位(>0.3 V)。
1.23V在电催化体系中的应用意义
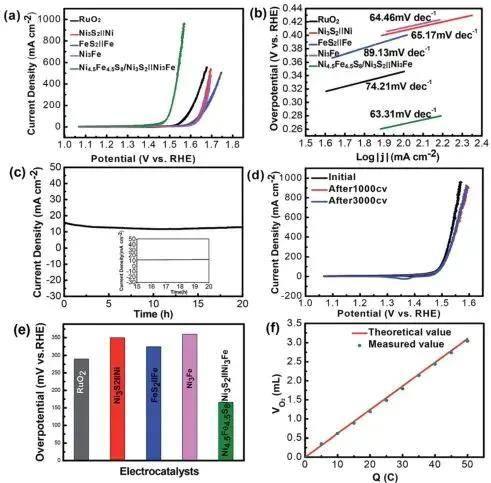
催化剂活性评估基准:
过电位(η)定义为η=E实际-1.23V(vs. RHE)。例如:商用IrO₂在10 mA/cm²时η≈300 mV;高效催化剂如NiFeOₓ可达η;Tafel斜率用于分析反应动力学机制。
电解槽设计依据:
理论最小电解电压为1.23 V(OER + HER),实际需1.8–2.2 V,能量效率由η决定。高温电解(如65°C)可优化反应速率,但需平衡材料稳定性。
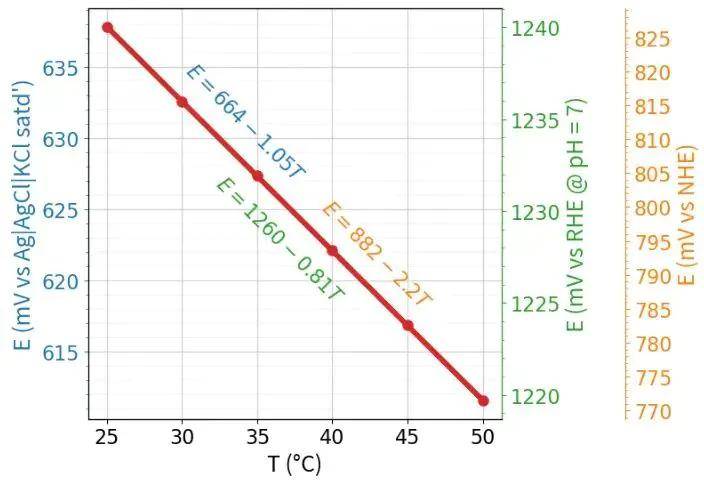
参考电极选择:
碱性体系常用Hg/HgO,酸性体系用Ag/AgCl,但数据需统一换算至RHE标度以保持可比性。
理想催化剂设计:需平衡中间体吸附能(如OH与OOH的ΔG差值≤0.2 eV),以逼近理论过电位极限0.37 V。
非热力学机制:晶格氧氧化机制(LOM)可能绕过传统吸附步骤,但伴随催化剂结构降解。
同位素效应:在D₂O中OER电势升至1.262 V vs. RDE,揭示质子转移对动力学的关键影响。
综上,1.23 V是OER反应的热力学基石,其精确性源于严格的标准条件定义和吉布斯自由能计算。实际应用中需结合动力学修正与环境参数调整,以指导高效电催化剂与电解系统的开发。