钙钛矿能做哪些计算?能带、态密度、差分电荷密度、bader电荷
钙钛矿的性能优化依赖于对其电子结构和材料特性的深入理解。
理论计算通过分析能带、态密度、功函数等电子结构参数,揭示了钙钛矿的光电转换机制;吸附能、d带中心和形成能的计算则为催化活性和材料稳定性提供了关键依据。此外,COOP/AIMD模拟从化学键合和动态行为角度,阐明了钙钛矿在高温或反应条件下的表现。
这些理论工具为钙钛矿电池的材料设计、界面优化和性能提升奠定了科学基础,推动了高效稳定钙钛矿器件的发展。
电子结构部分
能带/态密度
能带和态密度的计算对于研究钙钛矿具有至关重要的意义。它能够清晰地展现出钙钛矿材料中电子的能量分布情况,直接反映出材料的导电和光学性质。
通过分析能带的宽度、带隙位置以及导带和价带的结构,可以深入了解钙钛矿材料在光电转换过程中电子的激发和传输特性。
例如,带隙的大小决定了材料对光的吸收范围,合适的带隙宽度是实现高效光电转换的关键因素之一。同时,能带结构还能为材料的掺杂改性提供理论依据,指导研究人员通过调整材料的组成来优化其电子结构,从而提高电池的性能。
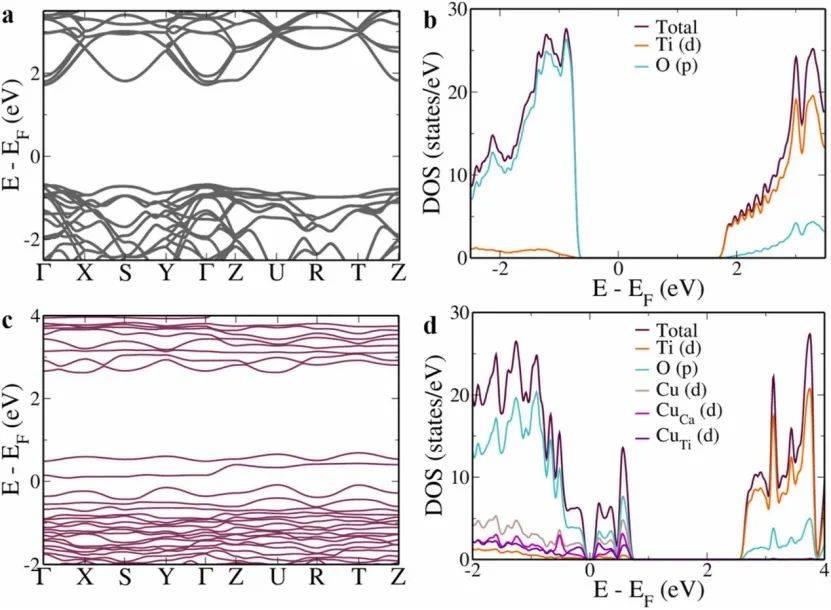
通过分析未掺杂CaTiO₃的能带结构发现其直接带隙为2.46 eV(Γ点),但由于DFT计算的局限性,这一数值可能存在低估。态密度分析表明,价带主要由O的2p轨道贡献,而导带则由Ti的3d轨道主导,这与SrTiO₃和BaTiO₃的电子行为一致。
铜掺杂后,带隙进一步减小,尤其是当Cu占据Ti位时,直接带隙降至1.98 eV,间接带隙更低(1.59 eV),显著提升了电荷分离效率,抑制了复合。这些结果为铜掺杂CaTiO₃在光催化应用中的优越性提供了电子层面的解释。
差分电荷密度/bader电荷
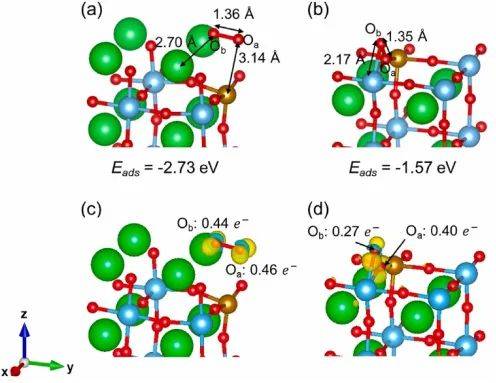
差分电荷密度和bader电荷的计算能够直观地展示钙钛矿材料中原子间的电荷转移和分布情况。它可以清晰地揭示出原子之间的成键特性,是离子键、共价键还是混合键,以及电荷在成键过程中的重新分配。
通过分析差分电荷密度,研究人员可以深入了解材料的化学 bonding 性质,判断原子之间的相互作用强弱,进而预测材料的稳定性和反应活性。在钙钛矿电池中,界面处的电荷转移效率对电池性能影响很大,差分电荷密度的计算可以帮助优化界面结构,提高电荷的分离和传输效率。
功函数
功函数的计算对于钙钛矿电池的电极界面设计至关重要。它表示材料表面逸出一个电子所需的最小能量,直接影响到材料与电极之间的电荷注入和提取效率。
通过调整钙钛矿材料的功函数,可以优化其与电极材料的能级匹配,减少界面处的能量势垒,提高电荷的传输效率,从而提升电池的整体性能。此外,功函数还与材料的表面化学性质相关,研究功函数的变化可以了解材料表面的修饰和缺陷对其电子发射能力的影响。
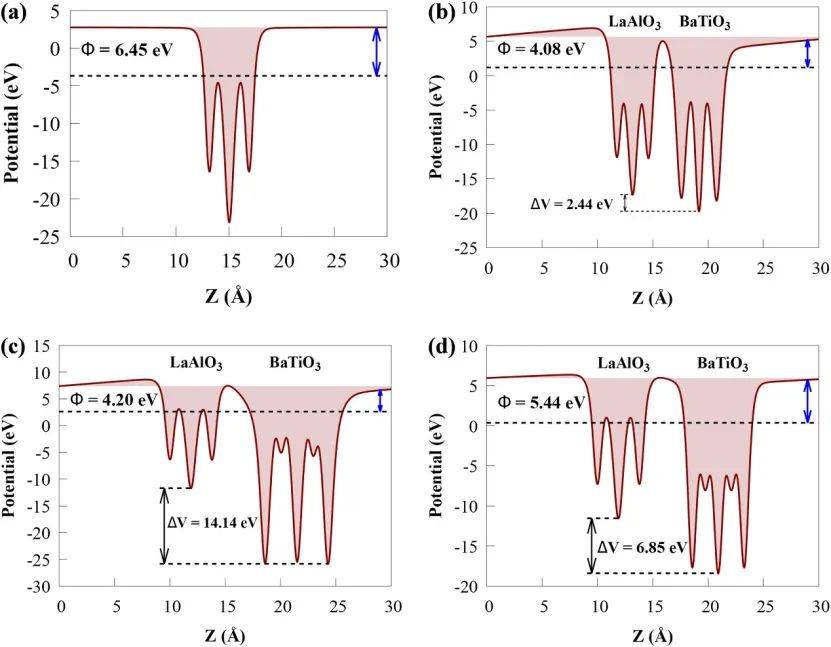
通过对比不同晶面(001)、(011)和(111)的功函数(分别为3.213 eV、5.282 eV和3.447 eV),发现(001)表面的功函数最低,表明其更易释放电子,与光催化活性增强的实验现象一致。
此外,异质结的功函数(如Conf(001)为4.12 eV)介于BaTiO₃和LaAlO₃(6.45 eV)之间,证实了界面处自发形成的内部电场能促进电荷分离,抑制电子-空穴复合。这一结果与能带对齐分析相结合,解释了异质结在可见光区吸收增强(带隙窄化至1.59-2.21 eV)和氧化还原电位(H₂O分解为H₂/O₂)优化的物理本质,为设计高效光催化剂提供了理论依据,尤其是(001)晶面因其低功函数和高活性位点密度成为最优选择。
ELF(电子局域函数)
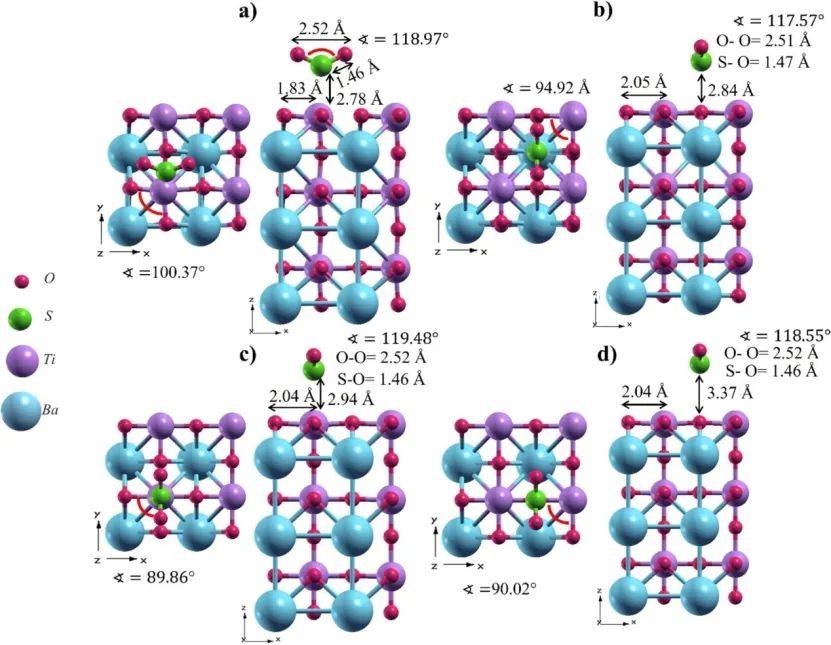
ELF 的计算能够直观地展示钙钛矿材料中电子的局域化程度,反映出电子在空间中的分布概率和化学键的性质。
通过分析 ELF,可以清晰地分辨出材料中的共价键、离子键和金属键区域,了解原子之间的成键类型和强度。这对于研究材料的晶体结构稳定性、相变过程以及缺陷形成机制具有重要意义。
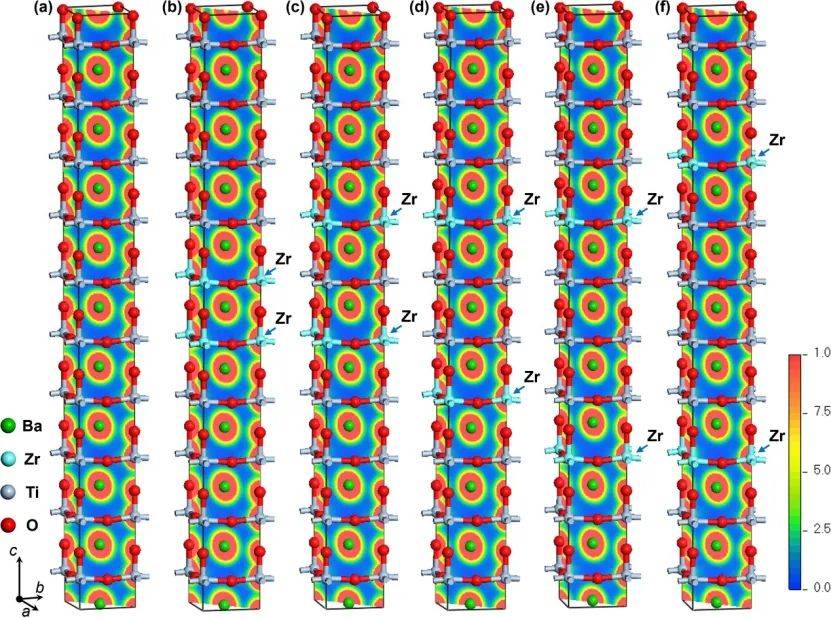
通过ELF计算揭示了BaTiO₃(BTO)和Zr掺杂BTO(BZT)体系中化学键的本质, Ba-O键呈现典型的离子键特征(ELF值低且电荷分布离散),而Ti-O和Zr-O键则表现为共价键(ELF值高且电子局域化明显),这与Mulliken布居分析的结果一致。
这种键合特性的差异直接影响材料的电子结构和稳定性:离子性的Ba-O键贡献于晶格刚性,而共价性的Ti/Zr-O键主导能带形成和载流子传输。
性能对比部分
吸附能
吸附能的计算在研究钙钛矿材料的表面催化反应和表面修饰过程中具有重要意义。
它表示分子或原子在材料表面吸附时的能量变化,反映了材料表面对吸附物种的结合能力。通过分析吸附能,可以确定哪些分子或原子容易吸附在钙钛矿表面,以及吸附过程对材料表面电子结构和化学性质的影响。
在钙钛矿电池中,表面吸附的物种可能会影响材料的光吸收、电荷分离和传输效率,例如空气中的水分和氧气可能会吸附在材料表面,导致材料的降解和性能下降。因此,研究吸附能可以指导设计表面保护层或修饰剂,提高材料的抗吸附能力和稳定性。
台阶图
台阶图(反应路径能量图)的计算能够展示钙钛矿材料在化学反应过程中的能量变化和反应路径。
它可以帮助研究人员了解化学反应的机理,确定反应的决速步骤和活化能,从而优化反应条件和材料结构。
在钙钛矿电池的制备过程中,涉及到多种化学反应,如薄膜的生长、界面的形成等,通过台阶图可以分析这些反应的热力学和动力学特性,指导制备工艺的优化,提高电池的制备效率和性能稳定性。此外,台阶图还可以用于研究材料的缺陷形成和演化过程,了解缺陷对材料性能的影响机制。
d 带中心
d 带中心的计算在研究过渡金属掺杂的钙钛矿材料的催化活性和电子结构调控方面具有重要意义。
对于含有过渡金属元素的钙钛矿材料,d 带中心的位置决定了材料的电子捐赠和接受能力,反映了材料的催化活性位点的电子状态。
通过调整 d 带中心的位置,可以优化材料对反应物的吸附和脱附能力,提高催化反应的效率。例如,在氧还原反应或氧析出反应中,d 带中心的合适位置可以使材料对氧分子的吸附既不过强也不过弱,从而实现高效的催化反应。此外,d 带中心还可以用于评估掺杂原子对材料电子结构的调控作用,为设计具有高催化活性的钙钛矿材料提供理论依据。
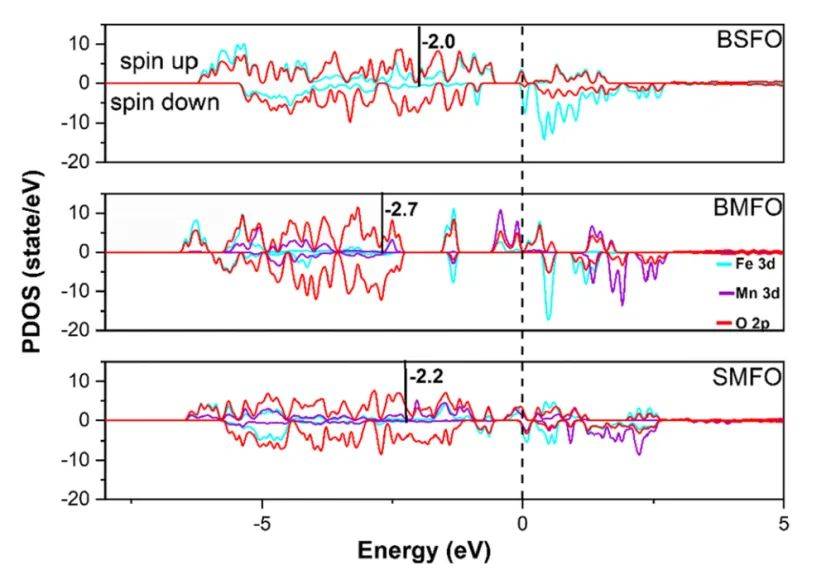
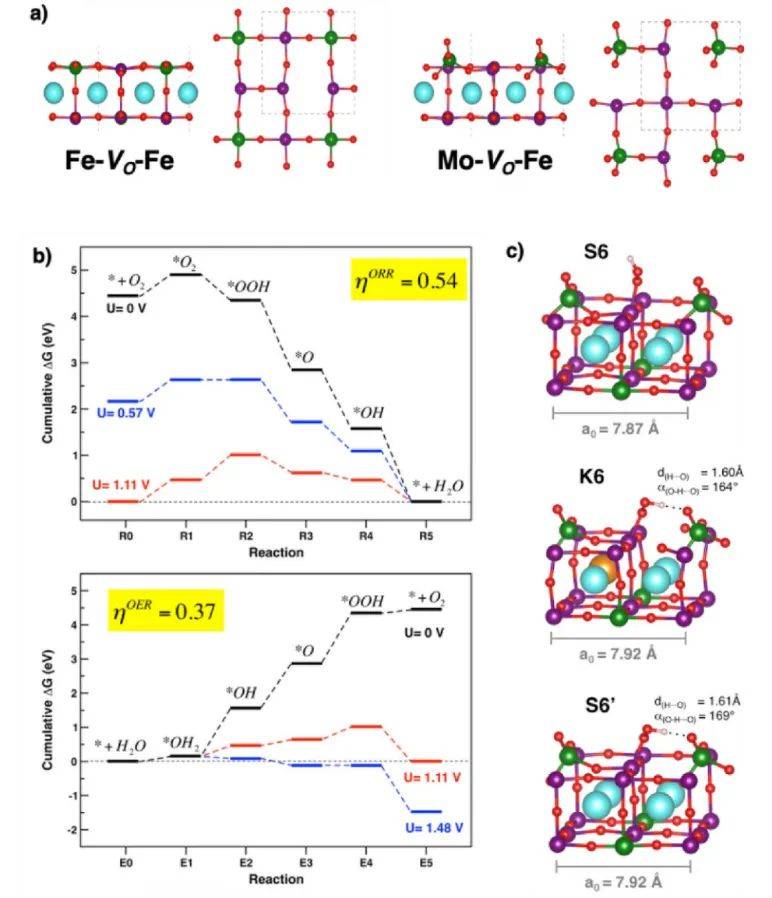
d带中心计算揭示了Ba0.5Sr0.5FeO3(BSFO)、SrMn0.5Fe0.5O3(SMFO)和BaMn0.5Fe0.5O3(BMFO)三种钙钛矿催化剂的电子结构与催化活性的关联。
通过分析过渡金属(Fe和Mn)的d轨道态密度(PDOS),发现BSFO的Fe 3d带中心最接近费米能级(黑色实线),表明其d电子更易参与表面反应,增强了氧空位形成能力(表5中Ev=1.07 eV)和氧化还原活性。
相比之下,SMFO和BMFO的d带中心位置较低,导致氧空位形成能更高(Ev分别为2.19 eV和4.06 eV),活性较弱。此外,Fe 3d轨道的自旋极化(自旋向上与向下态密度不对称)进一步优化了BSFO的电荷转移效率,
形成能
形成能的计算是评估钙钛矿材料稳定性的重要指标。它表示从单质或其他稳定化合物形成钙钛矿晶体时的能量变化,反映了材料在热力学上的稳定性。
通过分析形成能,可以确定不同组成和结构的钙钛矿材料的稳定性顺序,指导研究人员设计具有高稳定性的钙钛矿材料。例如,在高温、潮湿或光照条件下,形成能较低的钙钛矿材料更容易发生分解或相变,影响电池的使用寿命。因此,研究形成能可以帮助筛选出具有良好稳定性的材料组成和结构,为钙钛矿电池的实际应用提供保障。
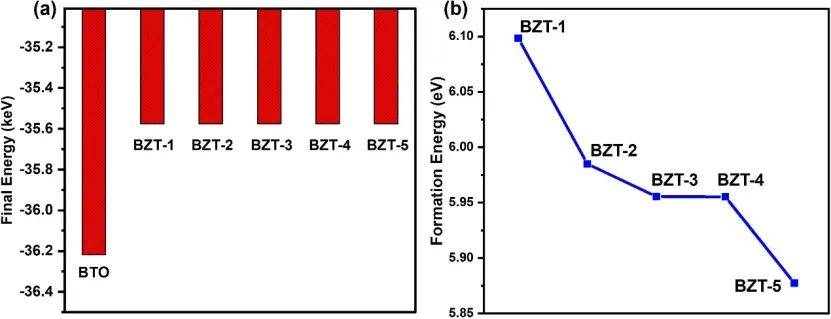
通过计算5种Zr掺杂BaTiO₃(BTO)体系的形成能从BZT-1(~6.09 eV)逐渐降低至BZT-5(~5.88 eV),表明随着Zr原子间距增大,体系的形成能减小,结构更易稳定存在。这一趋势与BZT-5的晶格参数和体积变化相吻合,说明Zr的分布方式显著影响材料的稳定性。
此外,形成能分析结合最终能量(进一步证实BZT-5是热力学最稳定的构型,这与其带隙值(1.71 eV)和电子结构的优化表现一致。这些结果为后续实验中选择低能耗、高稳定性的Zr掺杂策略奠定了基础。
个性化分析
过渡态
过渡态的计算在研究钙钛矿材料中的化学反应动力学过程中具有重要意义。
它能够确定化学反应过程中反应物转化为产物时所经过的能量最高的中间状态,即过渡态的结构和能量。
通过分析过渡态,可以了解化学反应的活化能和反应路径,揭示反应的机理和速率控制步骤。在钙钛矿电池中,涉及到许多关键的化学反应,如电荷的分离、传输和复合等过程,研究过渡态可以帮助优化材料的结构和组成,降低反应的活化能,提高反应的速率和效率,从而提升电池的性能。
COOP/COHP
COOP 和 COHP 的计算能够深入分析钙钛矿材料中原子之间的化学键合性质和强度。COOP 用于描述原子轨道之间的重叠程度和键合类型,COHP 则可以定量评估化学键的强弱和方向性。
通过分析 COOP 和 COHP,可以确定材料中原子之间的成键方式,是 σ 键、π 键还是其他类型的键,以及键的强弱和稳定性。这对于研究材料的晶体结构、力学性质和化学稳定性具有重要意义。
例如,在钙钛矿材料中,强的化学键可以提高材料的晶格稳定性,减少缺陷的形成,从而提高电池的使用寿命。此外,COOP 和 COHP 还可以用于研究掺杂和缺陷对材料化学键的影响,评估其对材料性能的调控作用。
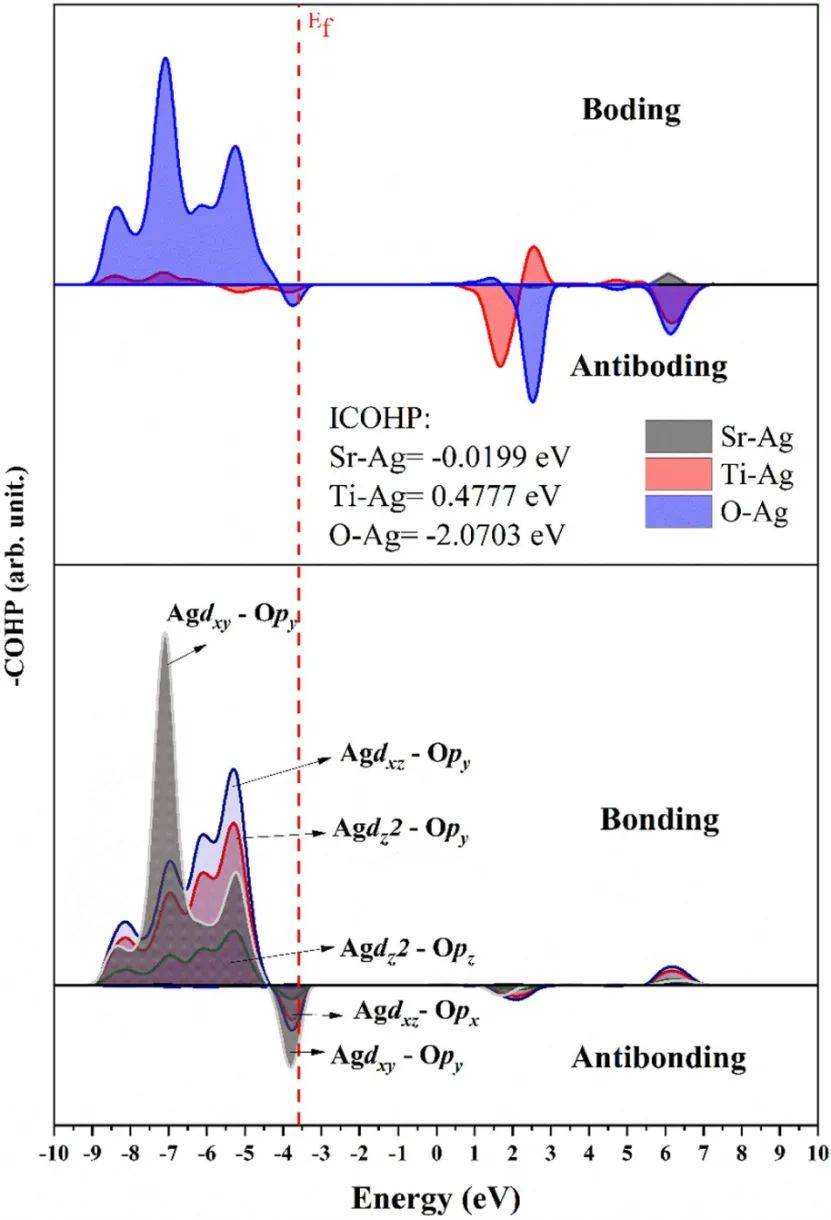
通过分析Ag-O轨道的相互作用,发现Ag的d轨道(dxy、dxz、dyz)与O的p轨道(px、py、pz)形成强σ型共价键,并在价带顶(VBM)区域产生显著的成键态),而导带底(CBM)则主要由[Ti-O]和[Ag-Ti]键贡献。
这一结果解释了Ag掺杂导致STO带隙从间接(R→Γ)转为直接(Γ→Γ)且减小0.15 eV的原因,同时证实了Ag抑制Sr电子态并引入新能级的关键作用。
COHP分析还表明,Ag-O键在VB内层存在强反键态,而在VB最大值处表现为强成键相互作用,这种独特的电子重分布优化了载流子分离效率,使Ag-STO成为p型半导体,显著提升了材料的光学性能(如光致发光和光吸收),为设计高效光催化与光电材料提供了理论依据。
AIMD(从头算分子动力学)
AIMD 的计算能够模拟钙钛矿材料在高温或动态环境下的原子运动和结构演化过程。
它可以研究材料的热稳定性、晶格动力学和缺陷扩散等行为,揭示材料在实际工作条件下的性能变化机制。通过 AIMD 模拟,可以观察到钙钛矿晶体在温度变化时的晶格畸变、原子位移和相变过程,评估材料的热稳定性和抗疲劳性能。
这对于设计能够在高温环境下稳定工作的钙钛矿电池具有重要意义。此外,AIMD 还可以用于研究材料中的载流子输运过程,了解原子运动对载流子迁移率的影响,为优化材料的电子传输性能提供理论支持。
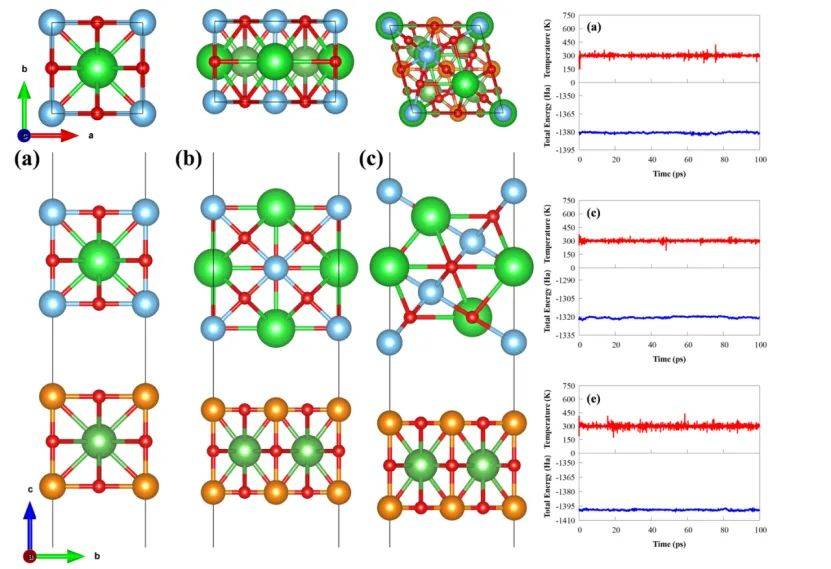
通过AIMD计算验证了BaTiO₃/LaAlO₃(001)异质结构在300K下的热力学稳定性,为材料在实际应用中的可靠性提供了关键依据。
通过分析能量和温度的波动幅度,AIMD模拟显示体系在300K下保持稳定波动(能量波动范围约0.5 eV,温度稳定在300K附近),表明异质结构在室温条件下无结构坍塌或原子扩散现象。这一结果与界面结合能)和平衡间距的计算相呼应,共同证实了(001)构型因最小的平衡间距(3.26 Å)和较强的界面相互作用而具有最优的稳定性。