
VASP 计算Bader电荷应用案例?
创始人
2025-11-07 15:35:24
0次
Bader电荷分析基于Richard Bader提出的一种原子电荷分析方法,通过电子密度的拓扑结构来划分原子区域,从而计算每个原子的电荷分布。深圳华算科技有限公司将详细介绍晶体氧化镁Bader电荷计算流程和数据处理方法。
结构优化
首先通过晶体结构数据库下载MgO的cif文件,并生成POSCAR文件,如下图所示。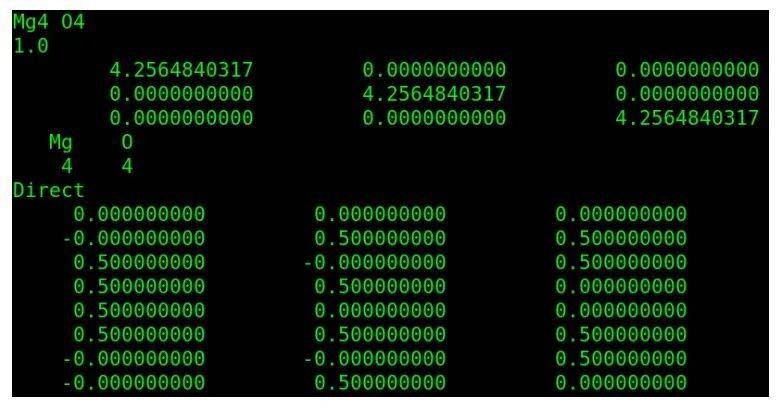
 然后编写结构优化的INCAR和KPOINTS文件。
然后编写结构优化的INCAR和KPOINTS文件。INCAR文件:
ISTART=0 #随机产生初始波函数
ICHARG=2 #从原子电荷密度产生体系初始电荷密度
PREC=M #计算精度为Medium
ISPIN=1 #关闭自旋极化
ALGO=N #DAV算法优化电子波函数
NELM=60 #电子波函数最多计算60步
EDIFF=1E-5 #电子波函数能量收敛标准1E-5 eV
ENCUT=400 #平面波截断能400 eV
IVDW=11 #考虑范德华力修正
IBRION=2 #共轭梯度法优化晶体结构和原子坐标
NSW=100 #晶体结构和原子坐标优化步数最大100步
ISIF=3 #优化原子坐标和晶体结构
EDIFFG=-0.1 #原子残余力小于0.1 eV/A
ISMEAR=0 #费米能级附近电子占据数为高斯分布
SIGMA=0.1 #高斯分布展宽0.1 eV
KPOINTS文件:
Automatic generation #注释行
0 #自动产生K点网格
G #布里渊区K点网格以
Gamma点为中心
5 5 5 #K点网格密度
0 0 0 #K点网格中心平移矢量
自洽计算
完成结构优化计算后,保持KPOINTS,POTCAR文件不变,将CONTCAR文件复制成POSCAR文件,并对结构优化的INCAR文件作如下修改:IBRION=-1 #固定结构和原子位置保持不变
NSW=0 #仅作一步自洽计算
LAECHG=.T. #计算Bader电荷
数据处理
完成自洽计算后,运行以下两个脚本,从而可以得到每个原子Bader电荷,它位于ACF.dat文件中CHARGE一列,其中1-4为Mg原子,5-8为O原子,排序和POSCAR一致。chgsum.pl AECCAR0 AECCAR2bader CHGCAR -ref CHGCAR_sum
相关内容
热门资讯
垂直计算申请运行时可重配置存储...
国家知识产权局信息显示,垂直计算有限公司申请一项名为“运行时可重配置存储器”的专利,公开号CN122...
苏州武乐川精密电子取得兼容多款...
国家知识产权局信息显示,苏州武乐川精密电子有限公司取得一项名为“一种新型兼容多款IC的运转料梭”的专...
引望申请供电电路及控制方法专利...
国家知识产权局信息显示,引望智能技术有限公司申请一项名为“供电电路、供电控制方法及相关装置”的专利,...
埃科光电(688610)7月2...
证券之星消息,截至2026年7月23日收盘,埃科光电(688610)报收于146.81元,下跌4.7...
上海玖陆易科技申请基于多传感器...
国家知识产权局信息显示,上海玖陆易科技发展有限公司申请一项名为“基于多传感器信息融合的智能疏水阀及其...
共青城市信宁智能制造研究院取得...
国家知识产权局信息显示,共青城市信宁智能制造研究院取得一项名为“基于摩擦纳米发电机的非接触式风速传感...
山东北方光学电子取得短波红外彩...
国家知识产权局信息显示,山东北方光学电子有限公司取得一项名为“一种短波红外彩色成像方法及光学系统”的...
美科物联取得电子把手锁专利
国家知识产权局信息显示,厦门美科物联科技有限公司取得一项名为“一种电子把手锁”的专利,授权公告号CN...
339亿!芯片大厂投了“大模型...
芯东西(公众号:aichip001) 编译 | ZeR0 编辑 | 漠影 芯东西7月23日报道,AM...
存储芯片集体涨价,手机也要跟着...
涨价潮背后,不只是存储的问题 —————————————— 近段时间,手机圈最热闹的话题不是某款新机...